Our Science
What is β-thalassemia?
Thalassemia is a serious single-gene genetic disease with a wide distribution in the world. It was first described by Thomas B. Cooley and Pearl Lee in 1925 in Italy as a form of severe anemia associated with splenomegaly and characteristic bone changes. As early diagnosed patients were mostly of Mediterranean origin, the disease was later termed as Thalassemia, from the Greek word for sea, thalassa. The hemoglobin tetramer is made of two α-globin chains or α-like (ζ)–globin chains and two β-globin chains or β-like(ε, γ, δ)–globin chains. Thalassemia is caused by an imbalance in hemoglobin synthesis due to the reduced production of at least one globulin polypeptide chain (β, α, γ, δ), and can be roughly divided into α-thalassemia and β-thalassemia [1]. β-Thalassemia is caused by reduced (β+) or absent(β0) synthesis of the β-globin, affecting the development and function of red blood cells, resulting in varying degrees of anemia. The clinical phenotypes are divided into three main categories according to the degree of impairment of β-globin production, namely β-thalassemia minor, β-thalassemia intermedia, and β-thalassemia major [2].
What are the symptoms of β-thalassemia?
β-Thalassemia minor occurs in heterozygotes (β/β+ or β/β0), and patients are usually asymptomatic or with mild to moderate microcytic anemia. A spectrum of clinical manifestations of β-thalassemia intermedia, which is between β-thalassemia minor and major, are caused by 2 abnormal-thalassemia alleles (β+/β0 or β+/β+). β-thalassemia major occurs in homozygotes (β0/β0) or severe cases of heterozygotes (β0/β+), resulting in severe β-globin deficiency, severe anemia, and bone marrow hyperactivity [3].
Children with β-thalassemia major are asymptomatic at birth. They begin to develop symptoms at 3-12 months of age, and may develop severe anemia add/or blood transfusion induced iron overload before 2 years old. Patients may present with jaundice, leg ulcers, and cholelithiasis, and often have an enlarged spleen. Due to active bone marrow hyperplasia, the cranial bones will change significantly after 1 year old, manifested as enlarged skull, bulging forehead, high cheekbones, collapsed bridge of the nose, and widened distance between the eyes, which is called "special face of thalassemia". The signs and symptoms become more apparent with age and the involvement of the long bones predisposes to pathological fractures, which in turn affect growth and development. In addition, myocardial iron overload may lead to heart failure, which is the consequence of myocardial damage caused by anemia and siderosis, and is one of the main causes of death in children; at the same time, hepatic siderosis leads to liver dysfunction and cirrhosis. Frequent blood transfusions can also increase the risk of concurrent transfusion-associated viral infections [4].
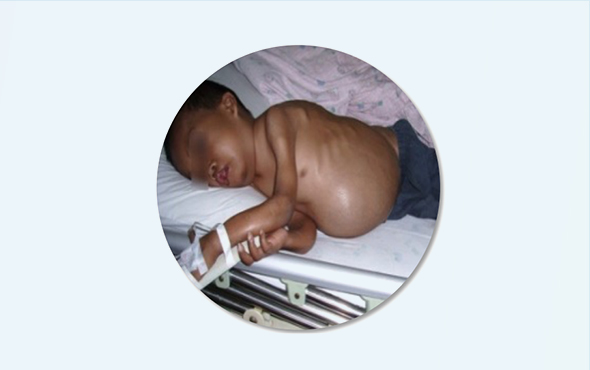
![Common β-thalassaemia complications[5] (Image source: 2021 Prevention and Control of Global Hemoglobinopathies-Chinese Version)](/uploadfiles/image/science/knowledge_02_02.jpg)
The epidemiology of β-thalassemia
The incidence of symptomatic thalassemia cases is estimated to be approximately 1 in 100,000 individuals in the general population. Originally, the disorder is particularly prevalent in South Asia, the Middle East, North Africa, and southern Europe. However, the prevalence of β‐thalassemia is increasing in other regions, including Northern Europe and North America, primarily due to population migration. About 1.5% of the world's population carries the β-thalassemia gene (80-90 million people), and at least tens of thousands of pediatrics patients with severe β-thalassemia are born every year, which has become a global public health problem [4]. According to the survey, the area with the most thalassemia gene carriers in China is Guangxi Province, and the carrier rate is as high as 20%. In China, β-thalassemia patients are mostly from the regions south of the Yangtze River, namely Guangdong Province, Guangxi Province, Hainan Province, Yunnan Province, and Guizhou Province. Blue Book of Thalassemia in China (2020) points out that the prevalence of thalassemia in Guangdong Province, Guangxi Province, and Hainan Province is from 9.2% to 24.07%, which is much higher compared to that of other provinces [6]. According to the "Blue Book of Thalassemia in China (2015)", there are about 30 million thalassemia gene carriers in China, and around 300,000 patients with thalassemia major and thalassemia intermedia who need to be treated. Among patients with thalassemia major and thalassemia intermedia, more than tens of thousands of patients are transfusion-dependent and the number of transfusion-dependent patients increases each year [7].
How do I know if I have β-thalassemia?
Patients with moderate and severe β-thalassemia usually develop typical clinical symptoms in childhood. Asymptomatic or mild patients may be detected incidentally via physical examination or other medical tests. Because thalassemia is hereditary, some people find out that they have thalassemia because they have relatives with thalassemia. The diagnosis of β-thalassemia major is mainly based on the following aspects [4]:
1)Clinical manifestations: Typical clinical features
1.1 Hematological changes:
1.1.1 Peripheral blood hemoglobin (Hb) < 60 g/L, microcytic hypochromic anemia, mean corpuscular volume (MCV) < 80fl, mean corpuscular hemoglobin (MCH) < 28pg, Mean erythrocyte hemoglobin concentration (MCHC)<320 g/L.
1.1.2 The bone marrow smear shows significant erythroid hyperplasia, with polychromatic and orthochromatic normoblast accounting for the majority, and the changes of mature erythrocytes were the same as those of peripheral blood.
1.1.3 The osmotic fragility of red blood cells was significantly reduced.
1.2 Hemoglobin electrophoresis: Hemoglobin electrophoresis at the first diagnosis shows that fetal hemoglobin (Hb F) is significantly increased, generally reaching 30%-90%, which is an important basis for the diagnosis of severe beta-thalassemia.
2)Regional and family surveys: Regional surveys show that the patients are from areas with high incidence of thalassemia. The parent of the child showed microcytic hypochromic anemia, and hemoglobin electrophoresis showed that the HbA2 content was elevated (3.5%-6.0%), and the Hb F was mostly normal.
3)Genetic diagnosis: Allele-specific oligonucleotide probe dot hybridization (PCR-ASO), reverse dot hybridization (RDB) and DNA sequencing methods can be used to detect the type of β-thalassemia gene defect. At present, more than 200 subtypes of β-globin gene mutations have been found in the world, and more than 50 subtypes have been found in the Chinese population. Therefore, the genetic defects of β-thalassemia are highly heterogeneous.
Can β-thalassemia be cured?
Patients with asymptomatic or beta-thalassemia minor do not require treatment. Patients with β-thalassemia intermedia should minimize blood transfusions to avoid other serious complications from iron overload. However, for patients with severe thalassemia (also called transfusion-dependent β-thalassemia, TDT), standardized lifelong blood transfusion and iron chelation are required, and the average annual treatment cost is more than $15,000 in developing country like China. Splenectomy or partial splenic artery embolization can be considered as palliative treatment. Hematopoietic stem cell transplantation is currently the only way to cure the disease, and the average medical cost is about $50,000. However, it also has disadvantages, which include difficulty in finding fully HLA-matched donors, high risk of graft-vs-host disease (GVHD), serious infectious complications caused by immunosuppressive therapy, and other side effects [7]. Regardless of the therapeutic strategies, β-thalassemia brings a heavy financial burden and pain to TDT patients and their families.
In recent years, gene therapy has made milestone progress in the treatment of TDT patients, which gives hope to cure thalassemia with this innovative approach. Gene therapy can overcome the shortcomings of traditional allogeneic hematopoietic stem cell transplantation. Gene therapy uses low intensity of myeloablative chemotherapy, has no risk of GVHD and do not require immunosuppressive therapy after transplantation. It is a very promising treatment method [8]. Hemogen is committed to the development and improvement of gene therapy, which makes curing thalassemia possible, by enhancing the efficacy and safety of the gene treatment and reducing the treatment cost.

Can β-thalassemia be prevented?
Prevention is an important measure to intervene thalassemia. Screening of gene carriers in the general population, prenatal diagnosis of high-risk pregnancies, and selective abortion to eliminate critically ill fetus are also the preferred measures, which are currently recognized by the international community, for the prevention of thalassemia. Population screening → carrier detection → genetic counseling for high-risk groups → prenatal diagnosis for high-risk pregnant women. This community-based population screening is now generally considered to be the most ideal mode of prevention [6].
At present, genetic screening is carried out in areas with high incidence of thalassemia in combination with premarital examination. For example, in Guangxi Province, if both or one of the couples are registered in Guangxi Province, "five free technical services" are provided, including free initial screening of thalassemia for pre-pregnancy couples and free prenatal diagnosis for high-risk pregnant women. Hainan Province fully implements free pre-marital examination, and thalassemia screening is listed as a mandatory item in pre-marital examination. With the advancement of thalassemia prevention projects, thalassemia has been effectively controlled in Guangdong Province, Guangxi Province and other provinces, and the birth rate of children with severe thalassemia has also declined year by year. According to the survey report of the " Blue Book of Thalassemia in China (2020)", after 2015, the number of newly diagnosed thalassemia patients born each year showed a slow downward trend [6]. In sum, β-thalassemia can be prevented with genetic screening and premarital examination.
References
[1] Bajwa H, Basit H. Thalassemia. 2021 Nov 5. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan–. PMID: 31424735.
[2] Origa R. β-Thalassemia. Genet Med. 2017 Jun;19(6):609-619. PMID: 27811859.
[3] Zakaria NA, Islam MA, Abdullah WZ, Bahar R, Mohamed Yusoff AA, Abdul Wahab R, Shamsuddin S, Johan MF. Epigenetic Insights and Potential Modifiers as Therapeutic Targets in β-Thalassemia. Biomolecules. 2021 May 18;11( 5): 755. PMID: 34070036.
[4] Editorial Board of Chinese Journal of Pediatrics, Hematology Group, Pediatric Branch of Chinese Medical Association. Guidelines for the diagnosis and treatment of β-thalassemia major (2017 edition). Chinese Journal of Pediatrics, 2018,56(10): 724-729.
[5] 2021 Prevention and Control of Global Hemoglobinopathies - Chinese Version
[6] China Philanthropy Research Institute, Beijing Normal University. Beijing Angel Mom Charity Foundation; China Siyuan Project Poverty Alleviation Foundation; China Thalassaemia Blue Book. (2020).
[7] China Philanthropy Research Institute, Beijing Normal University. Beijing Angel Mother Charity Foundation; China Siyuan Project Poverty Alleviation Foundation; China Thalassaemia Blue Book. (2015).
[8] Payen E. Efficacy and Safety of Gene Therapy for β-Thalassemia. N Engl J Med. 2022 Feb 3;386(5):488-490. PMID: 35108475.
